Normalization and Feature Selection
Step 03 / 08QC를 통해 low-quality cell을 제거한 뒤에는, 세포 간 sequencing depth 차이를 보정하고 downstream 분석에 중요한 variable genes를 선택하는 단계가 필요합니다. 이 단계는 PCA, clustering, annotation의 품질을 결정하는 핵심 과정입니다.
A) 왜 normalization이 필요한가?
Single-cell RNA-seq에서는 각 세포마다 sequencing depth와 library size가 다를 수 있습니다. 따라서 raw count를 그대로 비교하면, 실제 biological difference가 아니라 단순히 더 많이 시퀀싱된 세포가 더 높은 발현을 가진 것처럼 보일 수 있습니다.
Normalization의 목적은 이러한 기술적 차이를 줄이고, 세포 간 발현값을 비교 가능한 형태로 변환하는 것입니다. 즉, normalization을 통해 실제 biological signal에 더 가깝게 데이터를 해석할 수 있습니다.
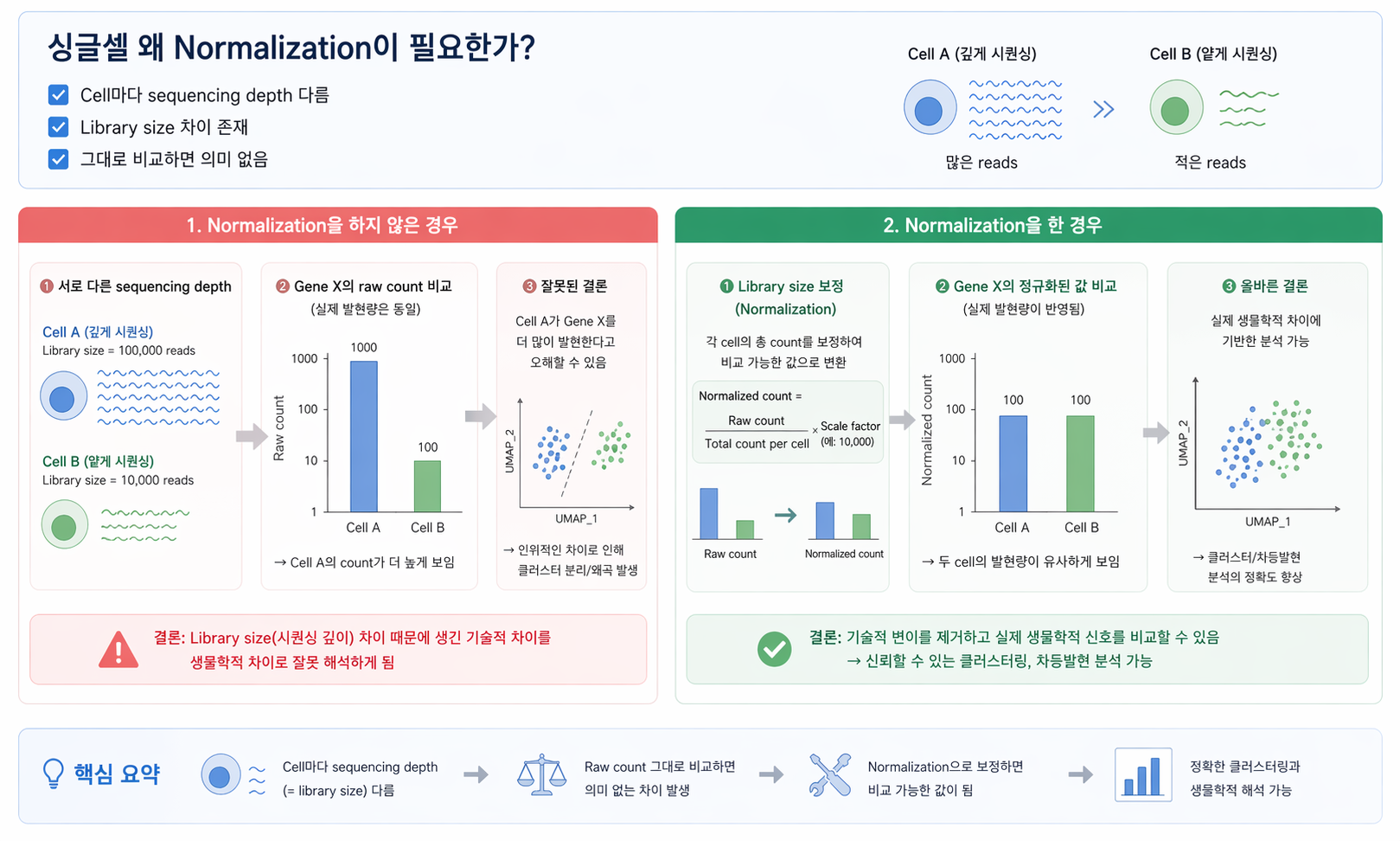
sequencing depth와 library size 차이를 보정하지 않으면 raw count 비교가 왜곡될 수 있습니다.
핵심 메시지
- Normalization은 세포 간 기술적 차이를 줄여 비교 가능한 값으로 바꾸는 과정입니다.
- 좋은 normalization은 clustering과 DEG 분석의 신뢰도를 높여줍니다.
B) NormalizeData vs SCTransform
Seurat에서 가장 자주 사용하는 정규화 방법은 NormalizeData()와
SCTransform()입니다.
NormalizeData()는 library size를 보정한 뒤 log transform을 적용하는
직관적이고 빠른 방법이며, 교육용 또는 기본 분석에 많이 사용됩니다.
반면 SCTransform()은 gene expression을 Negative Binomial model로 다루고,
sequencing depth와 같은 기술적 변동을 회귀 기반으로 보정합니다.
일반적으로 더 정교한 정규화 방법으로 여겨지지만, 계산량이 많고 개념적으로 더 복잡합니다.
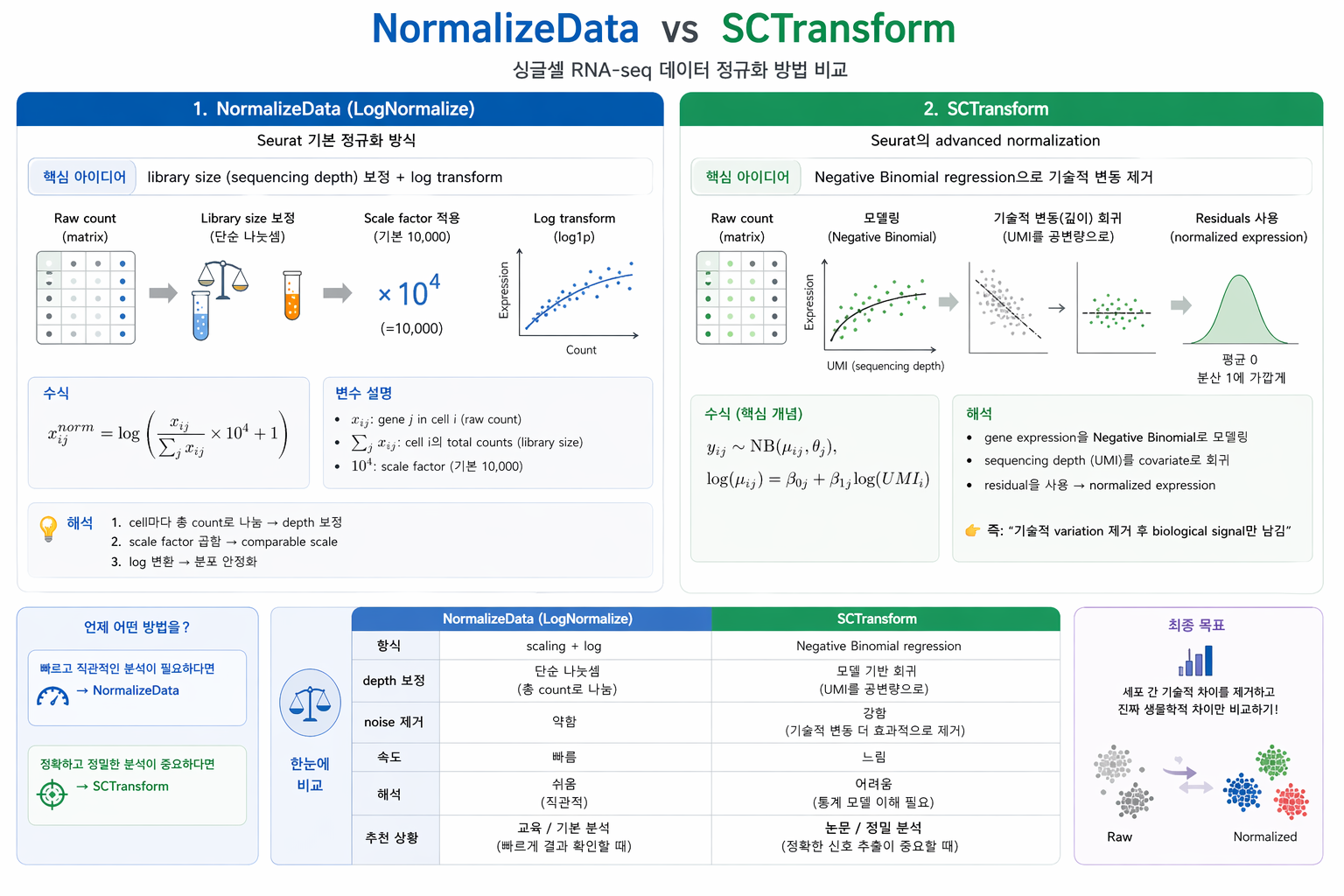
NormalizeData는 빠르고 직관적인 방식이며, SCTransform은 더 정교한 모델 기반 정규화 방법입니다.
| 항목 | NormalizeData | SCTransform |
|---|---|---|
| 핵심 방식 | library size 보정 + log transform | Negative Binomial regression 기반 정규화 |
| 장점 | 빠르고 직관적이며 교육용으로 적합 | 기술적 변동 제거 효과가 크고 더 정교함 |
| 단점 | noise 제거가 제한적일 수 있음 | 계산량이 많고 개념적으로 복잡함 |
| 추천 상황 | 기본 분석, 빠른 확인, 교육 목적 | 정밀 분석, 논문 수준 분석 |
본 튜토리얼에서는 먼저
NormalizeData()를 사용해 정규화 개념을 익히고,
SCTransform()은 이후 고급 단계에서 소개합니다.
C) Variable feature selection이 왜 중요한가?
모든 유전자를 그대로 downstream 분석에 사용하면, 변화가 거의 없는 housekeeping genes나 noise가 많은 유전자들이 함께 포함되어 PCA와 clustering 결과를 흐릴 수 있습니다.
따라서 세포 간 변동성이 큰 유전자들, 즉 highly variable genes (HVGs)를 선택하여 이후 차원 축소와 clustering에 사용하는 것이 일반적입니다. 이 과정은 biological signal을 더 잘 드러내고 계산 효율도 높여줍니다.
핵심 메시지
- Normalization은 비교 가능한 값을 만드는 과정입니다.
- Variable feature selection은 의미 있는 유전자를 선별하는 과정입니다.
- 이 두 단계가 잘 되어야 PCA와 clustering 품질도 좋아집니다.
D) NormalizeData 수행
본 실습에서는 6개 샘플 각각에 대해 NormalizeData()를 적용합니다.
sample_list <- lapply(sample_list, function(obj) {
NormalizeData(
obj,
normalization.method = "LogNormalize",
scale.factor = 10000
)
})
E) Variable feature selection 수행
정규화 후에는 각 샘플에서 변동성이 큰 유전자들을 선택합니다.
sample_list <- lapply(sample_list, function(obj) {
FindVariableFeatures(
obj,
selection.method = "vst",
nfeatures = 2000
)
})
F) Variable features 시각화
아래는 ND1 샘플에서 variable feature를 확인하는 예시입니다.
VariableFeaturePlot(sample_list[[1]])
상위 variable genes를 함께 표시하면 어떤 유전자들이 선택되었는지 더 쉽게 확인할 수 있습니다.
top10 <- head(VariableFeatures(sample_list[[1]]), 10)
LabelPoints(
plot = VariableFeaturePlot(sample_list[[1]]),
points = top10,
repel = TRUE
)
G) 모든 샘플에 대해 variable features 확인
실제 분석에서는 각 샘플마다 variable features의 분포가 다를 수 있으므로, 대표 샘플뿐 아니라 전체 샘플을 점검하는 것이 좋습니다.
for (nm in names(sample_list)) {
print(
VariableFeaturePlot(sample_list[[nm]]) + ggtitle(nm)
)
}
H) 다음 단계로 넘길 객체 준비
이후 integration 또는 clustering 단계에서는 정규화와 variable feature selection이 완료된 객체를 사용합니다.
names(sample_list)
head(VariableFeatures(sample_list[[1]]))
- Normalization은 세포 간 sequencing depth 차이를 줄여 비교 가능한 값을 만드는 단계입니다.
NormalizeData()는 빠르고 직관적이며,SCTransform()은 더 정교한 모델 기반 방법입니다.- Variable feature selection은 downstream 분석에 중요한 유전자들을 선별하는 과정입니다.
- 본 실습에서는 6개 mouse CRC 샘플에 대해 NormalizeData와 FindVariableFeatures를 적용했습니다.