Integration and Batch Correction
Step 04 / 08여러 샘플 또는 여러 배치에서 얻은 scRNA-seq 데이터는 batch effect의 영향을 받을 수 있습니다. Integration은 기술적 차이를 줄이고, 공통된 biological signal을 비교하기 위한 단계입니다.
A) 왜 Integration이 필요한가?
- 샘플이 서로 다른 날짜, 다른 라이브러리 preparation, 다른 operator, 다른 chemistry version에서 생성되면 기술적 차이가 생길 수 있습니다.
- 이 차이가 크면 실제 biological difference보다 batch effect가 더 크게 보일 수 있습니다.
- 이 경우 UMAP에서 샘플별로 분리되어 보이거나, 같은 cell type이 batch마다 따로 cluster될 수 있습니다.
핵심 포인트
- Integration은 biological difference를 없애는 과정이 아니라, 기술적 차이를 줄이는 과정입니다.
- 하지만 잘못 적용하면 실제 biological signal까지 희석할 수 있으므로 주의해야 합니다.
B) 언제 Integration을 고려해야 하나?
| 상황 | 설명 |
|---|---|
| 여러 환자/샘플 비교 | 서로 다른 샘플을 하나의 객체로 비교하려면 batch effect를 먼저 고려해야 합니다. |
| 실험 날짜가 다름 | 같은 조건이어도 날짜 차이, operator 차이, reagent 차이로 batch effect가 생길 수 있습니다. |
| 기술 플랫폼 차이 | 서로 다른 chemistry 또는 protocol에서 생성된 경우 integration이 더 중요해질 수 있습니다. |
| UMAP에서 샘플별 분리 | cell type보다 sample origin이 더 강하게 보이면 integration 필요성을 의심할 수 있습니다. |
C) 언제 조심해야 하나?
- 질환군과 대조군이 biological하게 매우 다를 때, 무리하게 integration하면 중요한 차이가 사라질 수 있습니다.
- 특정 샘플에만 존재하는 rare cell population이 integration 과정에서 희석될 수 있습니다.
- 따라서 integration 전후 결과를 모두 비교하면서 해석해야 합니다.
D) Seurat Integration 기본 흐름
Seurat에서는 각 샘플을 개별적으로 전처리한 뒤, anchor를 찾고 데이터를 통합합니다.
library(Seurat)
# sample list 예시
obj.list <- list(sample1 = obj1, sample2 = obj2, sample3 = obj3)
# 각 샘플에서 normalization 및 variable features
obj.list <- lapply(obj.list, function(x) {
x <- NormalizeData(x)
x <- FindVariableFeatures(x, selection.method = "vst", nfeatures = 2000)
return(x)
})
# integration features 선택
features <- SelectIntegrationFeatures(object.list = obj.list)
# anchor 찾기
anchors <- FindIntegrationAnchors(
object.list = obj.list,
anchor.features = features
)
# 데이터 통합
obj.integrated <- IntegrateData(anchorset = anchors)
E) 통합 후 기본 분석
통합된 객체에서는 보통 integrated assay를 기준으로 scaling, PCA, UMAP, clustering을 수행합니다.
DefaultAssay(obj.integrated) <- "integrated"
obj.integrated <- ScaleData(obj.integrated, verbose = FALSE)
obj.integrated <- RunPCA(obj.integrated, npcs = 30, verbose = FALSE)
obj.integrated <- RunUMAP(obj.integrated, dims = 1:20)
obj.integrated <- FindNeighbors(obj.integrated, dims = 1:20)
obj.integrated <- FindClusters(obj.integrated, resolution = 0.5)
F) Harmony 기반 보정 예시
Harmony는 PCA 공간에서 batch effect를 줄이는 접근입니다. Seurat integration과 함께 자주 비교되는 방법입니다.
library(Seurat)
library(harmony)
obj <- NormalizeData(obj)
obj <- FindVariableFeatures(obj)
obj <- ScaleData(obj)
obj <- RunPCA(obj, npcs = 30)
obj <- RunHarmony(obj, group.by.vars = "sample")
obj <- RunUMAP(obj, reduction = "harmony", dims = 1:20)
obj <- FindNeighbors(obj, reduction = "harmony", dims = 1:20)
obj <- FindClusters(obj, resolution = 0.5)
G) Integration 전후 비교
Integration이 잘 되었는지는 반드시 전후 UMAP을 비교해서 확인해야 합니다.
# integration 전
DimPlot(obj, group.by = "sample")
# integration 후
DimPlot(obj.integrated, group.by = "sample")
DimPlot(obj.integrated, label = TRUE)
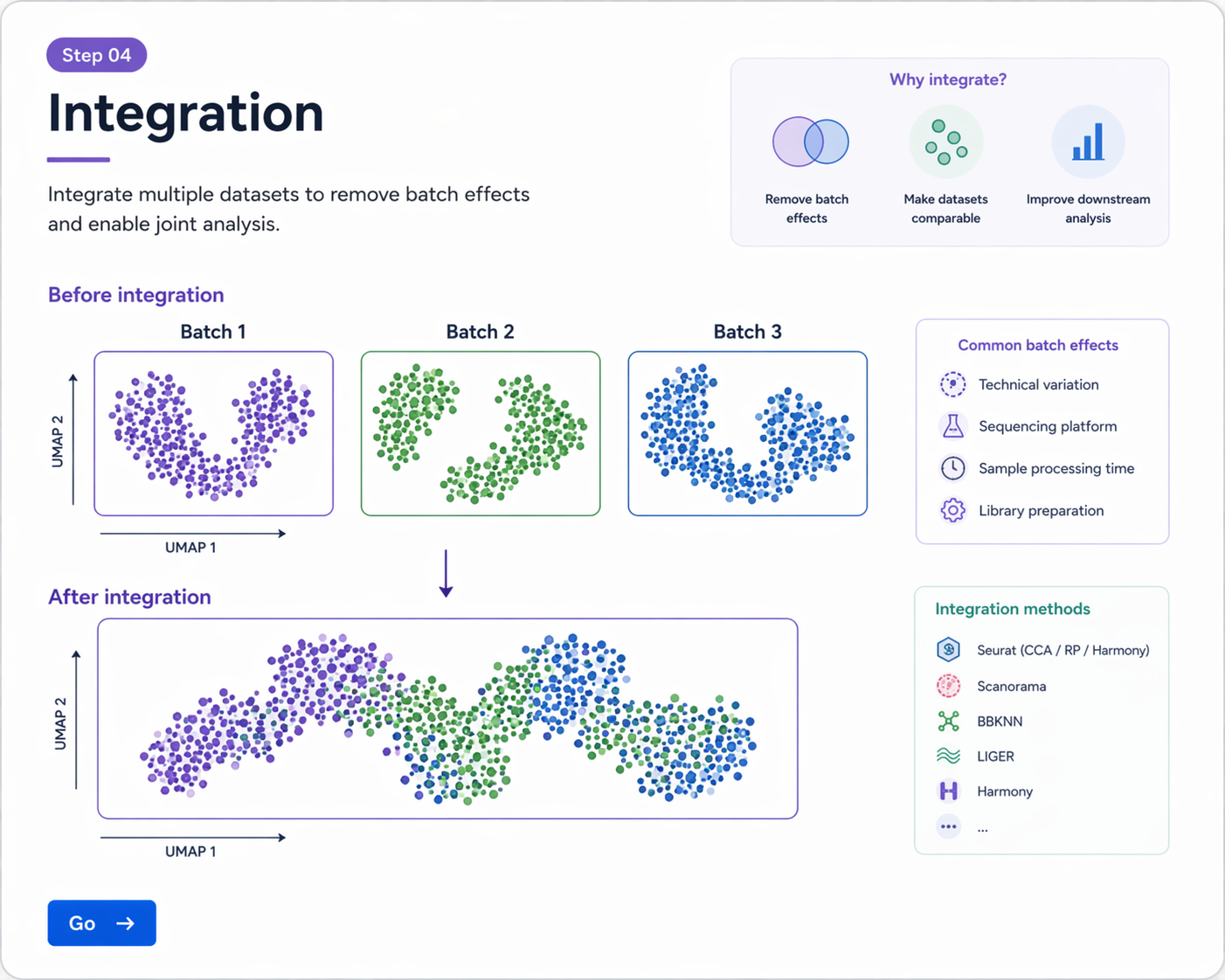
Integration은 서로 다른 batch 또는 sample 간 기술적 차이를 줄여, 공통된 cell type 구조를 더 잘 비교할 수 있도록 돕습니다.
H) 해석 시 주의할 점
- batch effect가 줄어들었다고 해서 무조건 좋은 결과는 아닙니다.
- 실제 biological difference까지 없어졌는지 함께 확인해야 합니다.
- cell type marker, sample distribution, known biology를 함께 점검해야 합니다.
- 분석 목적이 “통합 atlas 구축”인지, “조건 차이 비교”인지에 따라 integration 전략이 달라질 수 있습니다.
실전 팁
- 항상 integration 전 raw/standard workflow 결과도 함께 보관하세요.
- UMAP만 보지 말고, marker gene가 자연스럽게 유지되는지 확인해야 합니다.
- 샘플 간 mixing이 좋아 보여도, rare population이 사라지지 않았는지 확인하는 것이 중요합니다.
I) 실습용 전체 코드 예시
library(Seurat)
# 1. 개별 샘플 전처리
obj.list <- list(sample1 = obj1, sample2 = obj2)
obj.list <- lapply(obj.list, function(x) {
x <- NormalizeData(x)
x <- FindVariableFeatures(x, selection.method = "vst", nfeatures = 2000)
return(x)
})
# 2. integration features 선택
features <- SelectIntegrationFeatures(object.list = obj.list)
# 3. anchors 찾기
anchors <- FindIntegrationAnchors(
object.list = obj.list,
anchor.features = features
)
# 4. integrate
obj.integrated <- IntegrateData(anchorset = anchors)
# 5. downstream analysis
DefaultAssay(obj.integrated) <- "integrated"
obj.integrated <- ScaleData(obj.integrated)
obj.integrated <- RunPCA(obj.integrated, npcs = 30)
obj.integrated <- RunUMAP(obj.integrated, dims = 1:20)
obj.integrated <- FindNeighbors(obj.integrated, dims = 1:20)
obj.integrated <- FindClusters(obj.integrated, resolution = 0.5)
# 6. visualization
DimPlot(obj.integrated, group.by = "sample")
DimPlot(obj.integrated, label = TRUE)
요약
- Integration은 여러 샘플/배치의 기술적 차이를 줄이기 위한 단계입니다.
- Seurat integration은 anchor 기반으로 공통 구조를 찾고 데이터를 통합합니다.
- Harmony는 PCA 공간에서 batch effect를 보정하는 대안적 방법입니다.
- integration 전후를 반드시 비교하고, biological signal 손실 여부를 함께 확인해야 합니다.