Step 02. Adapter Trimming (Cutadapt)
페어드엔드 FASTQ(R1/R2)에서 Illumina 어댑터 서열을 제거합니다. 기본 쓰레드는 20을 사용합니다.
자세한 사용법은 공식 문서(Cutadapt User Guide)를 참고하세요.
0) 시작하기 전에
Conda 환경을 활성화하면 터미널 프롬프트 앞에 보통 (wgs_env)처럼 환경명이 표시됩니다.
# Conda 환경 활성화
conda activate wgs_env
# 프롬프트 예시 (환경명이 괄호로 표시됨)
# (wgs_env) kang@host:~/path/to/fastq$
1) 입력 데이터 & 어댑터 서열
- 페어드엔드(PE) 샘플은 파일이 2개로 구성됩니다:
SAMPLE_R1.fastq.gz(Forward/Read1),SAMPLE_R2.fastq.gz(Reverse/Read2) - 일반적인 Illumina TruSeq 어댑터:
- R1 어댑터(Forward):
AGATCGGAAGAGCACACGTCTGAACTCCAGTCA - R2 어댑터(Reverse):
AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT
- R1 어댑터(Forward):
원한다면 압축 FASTQ에서 어댑터 서열이 보이는지 간단히 확인할 수 있습니다(빠른 스폿 체크).
# R1에서 Forward 어댑터 검색 (gzip 압축 파일에는 zgrep 권장)
zgrep -a 'AGATCGGAAGAGCACACGTCTGAACTCCAGTCA' sample1_1.fastq.gz | head
# R2에서 Reverse 어댑터 검색
zgrep -a 'AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT' sample1_2.fastq.gz | head
# 대화형으로 보고 싶다면:
# zless sample1_1.fastq.gz # 연 후에 /AGATCGG... 로 검색
2) 변수 설정
# 공통 변수
THREADS=20
OUTDIR="/home/kang/God_Nas/WGS_2nd/Trimmed_fastq"
mkdir -p "${OUTDIR}"
# 어댑터 서열
ADAPTER_FWD="AGATCGGAAGAGCACACGTCTGAACTCCAGTCA"
ADAPTER_REV="AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT"
3) 배치 실행 스크립트 (run_cutadapt.sh)
현재 폴더의 *_R1.fastq.gz를 기준으로 페어(R2)를 찾아 일괄 처리합니다.
#!/usr/bin/env bash
set -euo pipefail
THREADS=20
OUTDIR="/home/kang/God_Nas/WGS_2nd/Trimmed_fastq"
mkdir -p "${OUTDIR}"
ADAPTER_FWD="AGATCGGAAGAGCACACGTCTGAACTCCAGTCA"
ADAPTER_REV="AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT"
shopt -s nullglob
for R1 in *_R1.fastq.gz; do
SAMPLE="$(basename "${R1}" _R1.fastq.gz)"
R2="${SAMPLE}_R2.fastq.gz"
if [[ ! -f "${R2}" ]]; then
echo "[WARN] ${SAMPLE}: paired R2 not found. Skip." >&2
continue
fi
echo "[Cutadapt] ${SAMPLE}"
cutadapt -j "${THREADS}" \
-a "${ADAPTER_FWD}" \
-A "${ADAPTER_REV}" \
-o "${OUTDIR}/${SAMPLE}_R1.trimmed.fastq.gz" \
-p "${OUTDIR}/${SAMPLE}_R2.trimmed.fastq.gz" \
"${R1}" "${R2}"
done
echo "All trimming jobs finished. Output: ${OUTDIR}"
4) 실행
# 트리밍할 FASTQ들이 있는 폴더로 이동
cd /path/to/fastq_folder
# (wgs_env) 프롬프트 확인 후 실행
chmod +x run_cutadapt.sh
bash run_cutadapt.sh
5) 출력물
${OUTDIR}/SAMPLE_R1.trimmed.fastq.gz${OUTDIR}/SAMPLE_R2.trimmed.fastq.gz
6) (옵션) 로그 저장 & 품질/길이 필터
# 로그 저장 예시: 각 샘플의 실행 리포트를 파일로 남김
cutadapt -j "${THREADS}" \
-a "${ADAPTER_FWD}" -A "${ADAPTER_REV}" \
-o "${OUTDIR}/${SAMPLE}_R1.trimmed.fastq.gz" \
-p "${OUTDIR}/${SAMPLE}_R2.trimmed.fastq.gz" \
"${R1}" "${R2}" > "${OUTDIR}/${SAMPLE}.cutadapt.log" 2>&1
# (선택) 추가 옵션 예시
# -q 20 : Q20 미만의 말단 염기 트리밍
# -m 30 : 길이 30bp 미만 리드는 버림
# 예) cutadapt ... -q 20 -m 30 ...
7) 품질 확인 (FastQC 전/후 비교)
트리밍 전/후로 FastQC를 실행해 품질 변화를 확인합니다. 여러 샘플의 리포트를 한 번에 모아보려면 MultiQC를 사용합니다. FastQC 문서: FastQC Home
실행 명령
# (전) 원본 FASTQ
mkdir -p qc_raw
fastqc -t 20 *.fastq.gz -o qc_raw
# (후) 트리밍 FASTQ
mkdir -p qc_trimmed
fastqc -t 20 "${OUTDIR}"/*trimmed.fastq.gz -o qc_trimmed
# (요약) MultiQC로 한 페이지에 모으기
mkdir -p qc_multiqc
multiqc qc_raw qc_trimmed -o qc_multiqc
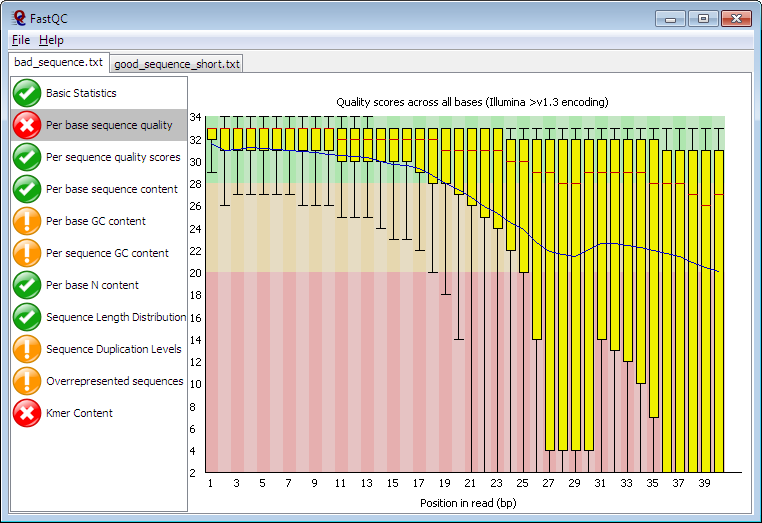
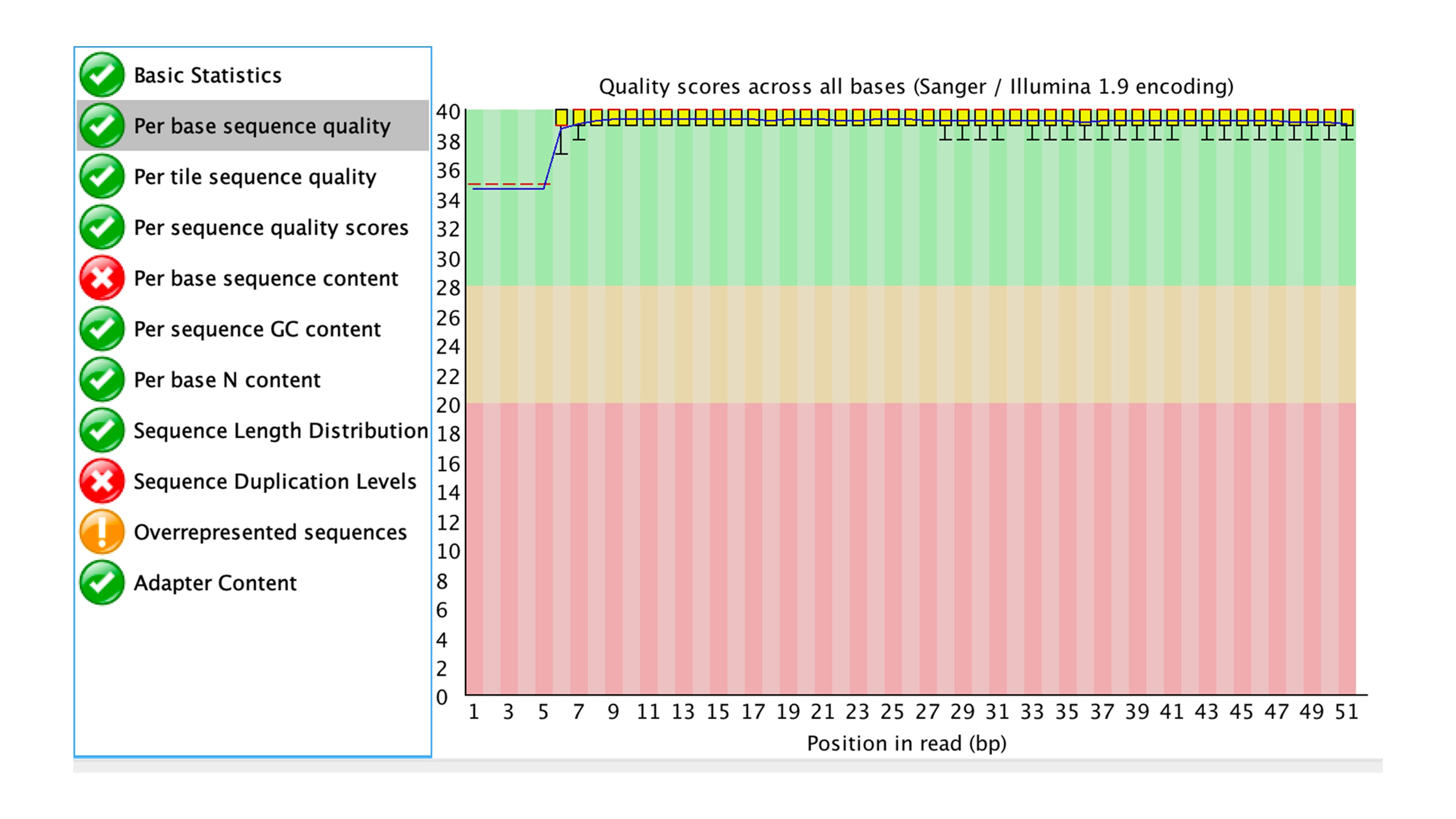
Per base sequence quality (전/후 비교)
그래프의 y축은 Phred 품질 점수(Q)이며, Q = -10·log10(p) (p: 오류확률)입니다.
Q20≈1% 오류(99% 정확), Q30≈0.1% 오류(99.9% 정확)로 일반적으로 Q30 이상이면 양호합니다.
배경색은 FastQC 권고: 초록(좋음) / 노랑(보통) / 빨강(나쁨).
- 인간 WGS 30× 기준: 약
3×109 bp × 30 = 9×1010bases - Q20(1% 오류) → 예상 에러 ≈
0.01 × 90G = 0.9G(약 9억) - Q30(0.1% 오류) → 예상 에러 ≈
0.001 × 90G = 90M(약 9천만)
동일 데이터라도 에러가 10배 차이 → 정렬 품질, 변이콜링 위양성/저빈도 변이 탐지에 큰 영향. 그래서 실무에선 “대부분 Q30 이상 + 말단 트리밍”을 기본으로 사용합니다.
Before trimming
After trimming
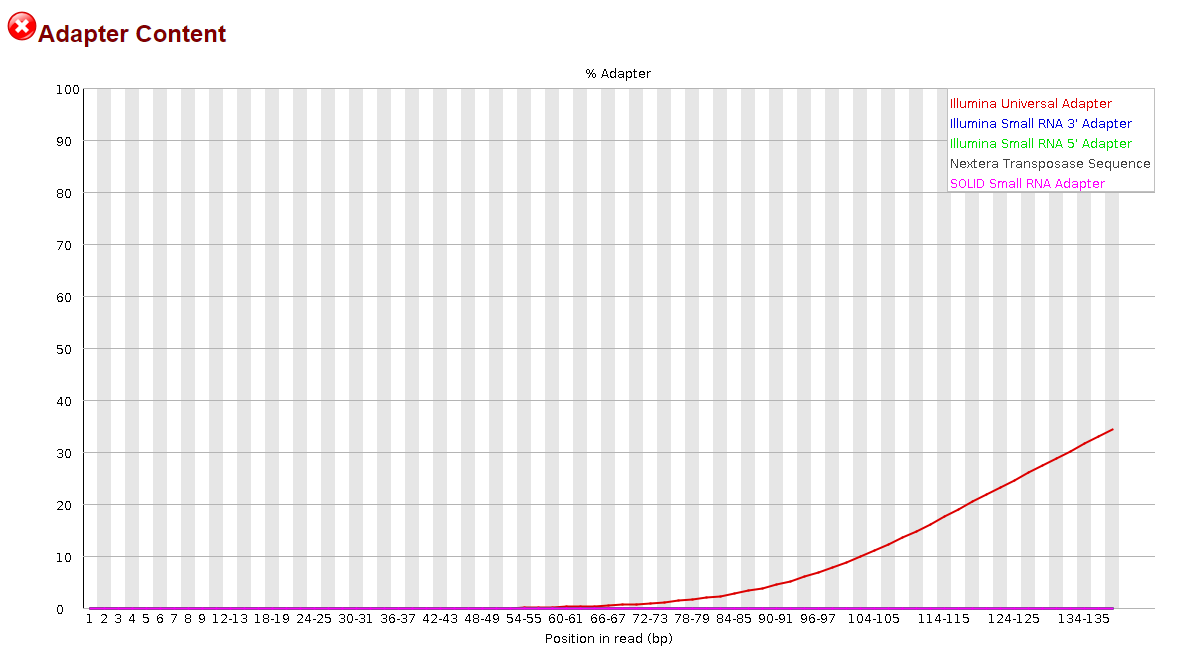
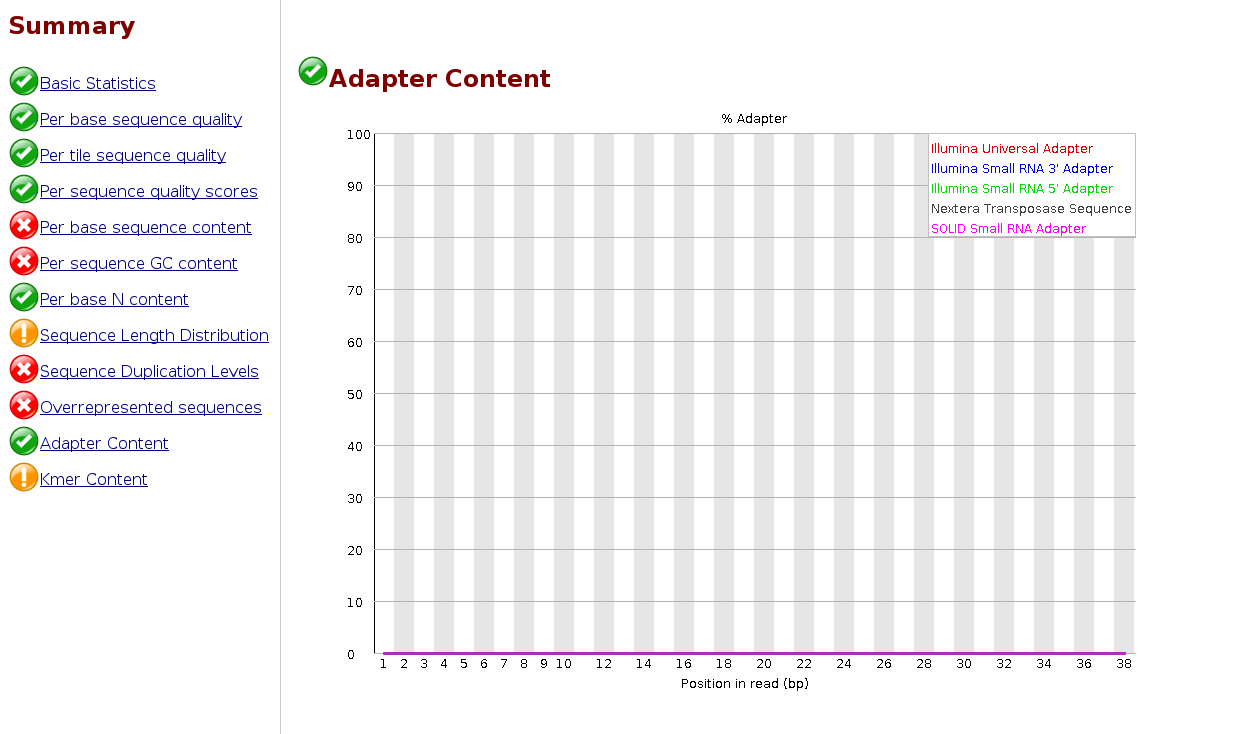
Adapter Content 확인 (전/후 비교)
FastQC의 Adapter Content 모듈에서 위치별 어댑터 잔존 비율을 확인할 수 있습니다. 일반적으로 트리밍 전엔 특정 위치 이후 어댑터 비율이 상승하고, 트리밍 후엔 0에 가깝게 떨어지는 것이 이상적입니다.
Before trimming
After trimming
-q)이나 길이 필터(-m)를 적용하세요.