DEG and Pathway Analysis
Step 07 / 08Annotation 이후에는 cluster 간 또는 조건 간 differentially expressed genes (DEGs)를 확인하고, pathway 분석을 통해 해당 세포 집단의 biological meaning을 해석할 수 있습니다.
A) DEG 분석이란?
- DEG 분석은 두 집단 사이에서 유의하게 차이나는 유전자를 찾는 과정입니다.
- 예를 들어, T cell과 B cell을 비교하거나, 같은 cell type 내에서 control과 disease를 비교할 수 있습니다.
- 단순 marker gene 확인보다 한 단계 더 나아가, 어떤 biological process가 활성화되어 있는지 해석할 수 있습니다.
핵심 포인트
- cluster 간 비교와 condition 간 비교는 목적이 다릅니다.
- 비교 대상이 명확해야 해석도 명확해집니다.
B) Cluster 간 DEG 분석
Seurat에서는 FindMarkers()를 이용해 두 cluster 간 차이를 비교할 수 있습니다.
deg_cluster <- FindMarkers(
obj,
ident.1 = "T cells",
ident.2 = "B cells",
min.pct = 0.25,
logfc.threshold = 0.25
)
head(deg_cluster)
결과 테이블에는 p-value, adjusted p-value, average log2 fold change, 발현 비율 등이 포함됩니다.
C) Condition 간 DEG 분석
같은 cell type 내에서 조건 간 비교를 하고 싶다면, 먼저 특정 세포 유형만 subset한 뒤 비교할 수 있습니다.
tcell_obj <- subset(obj, idents = "T cells")
Idents(tcell_obj) <- "condition"
deg_condition <- FindMarkers(
tcell_obj,
ident.1 = "disease",
ident.2 = "control",
min.pct = 0.25,
logfc.threshold = 0.25
)
head(deg_condition)
D) 상위 DEG 확인
상위 유전자를 정리하면 cluster 또는 condition을 구분하는 핵심 유전자를 빠르게 확인할 수 있습니다.
library(dplyr)
deg_cluster %>%
rownames_to_column("gene") %>%
arrange(desc(avg_log2FC)) %>%
head(20)
E) Heatmap 시각화
DEG 결과 중 상위 marker 또는 상위 DEG를 heatmap으로 시각화하면 group 간 차이를 직관적으로 확인할 수 있습니다.
top_genes <- rownames(deg_cluster)[1:20]
DoHeatmap(obj, features = top_genes) + NoLegend()
F) Volcano plot 예시
Volcano plot은 fold change와 statistical significance를 함께 보여주는 대표적 시각화입니다.
library(ggplot2)
library(tibble)
volcano_df <- deg_cluster %>%
rownames_to_column("gene") %>%
mutate(
significant = ifelse(p_val_adj < 0.05 & abs(avg_log2FC) > 0.5, "Yes", "No")
)
ggplot(volcano_df, aes(x = avg_log2FC, y = -log10(p_val_adj), color = significant)) +
geom_point(size = 1.2) +
theme_classic() +
labs(x = "avg_log2FC", y = "-log10 adjusted p-value")
G) Violin plot으로 개별 유전자 확인
VlnPlot(
obj,
features = c("CD3D", "MS4A1", "EPCAM"),
group.by = "celltype",
ncol = 3
)
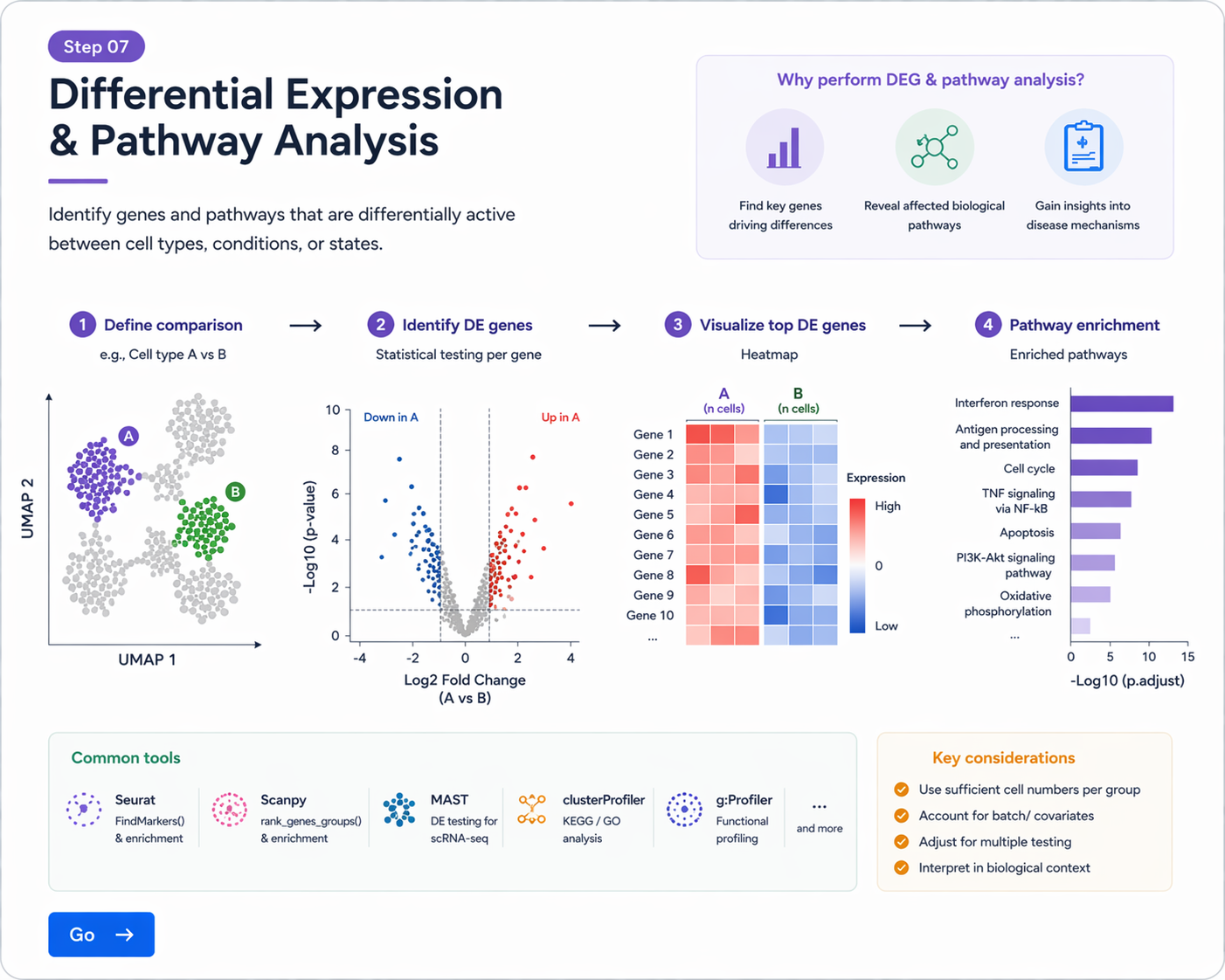
DEG 분석으로 차이를 보이는 유전자를 찾고, pathway 분석을 통해 biological function을 해석할 수 있습니다.
H) GO / KEGG enrichment 분석
DEG 리스트를 이용하면 enrichment 분석을 통해 활성화된 biological process를 확인할 수 있습니다.
library(clusterProfiler)
library(org.Hs.eg.db)
sig_genes <- deg_cluster %>%
rownames_to_column("gene") %>%
filter(p_val_adj < 0.05, avg_log2FC > 0.5) %>%
pull(gene)
gene_df <- bitr(sig_genes,
fromType = "SYMBOL",
toType = "ENTREZID",
OrgDb = org.Hs.eg.db)
ego <- enrichGO(
gene = gene_df$ENTREZID,
OrgDb = org.Hs.eg.db,
ont = "BP",
pAdjustMethod = "BH",
readable = TRUE
)
head(ego)
dotplot(ego)
I) GSEA 예시
ORA뿐 아니라 전체 ranked gene list를 사용하는 GSEA도 자주 사용됩니다.
gene_list <- deg_cluster$avg_log2FC
names(gene_list) <- rownames(deg_cluster)
gene_list <- sort(gene_list, decreasing = TRUE)
gsea_res <- gseGO(
geneList = gene_list,
OrgDb = org.Hs.eg.db,
ont = "BP",
keyType = "SYMBOL",
verbose = FALSE
)
head(gsea_res)
ridgeplot(gsea_res)
J) 실전 해석 포인트
- 유의한 p-value만 보지 말고, effect size (log fold change)도 함께 봐야 합니다.
- 세포 수가 많으면 작은 차이도 매우 유의하게 나올 수 있으므로 biological relevance를 함께 고려해야 합니다.
- DEG는 annotation을 보강할 수도 있고, condition-specific state를 설명할 수도 있습니다.
- Pathway 결과는 해석을 돕지만, gene set database의 특성과 bias도 염두에 두어야 합니다.
실전 팁
- 비교 그룹을 너무 넓게 잡기보다, biological question에 맞게 명확히 정의하세요.
- condition 비교는 가능하면 같은 cell type 내에서 수행하는 것이 해석이 더 명확합니다.
- heatmap, violin plot, enrichment 결과를 함께 제시하면 설명력이 높아집니다.
K) 전체 실습 코드 예시
library(Seurat)
library(dplyr)
library(tibble)
library(clusterProfiler)
library(org.Hs.eg.db)
# 1. cluster 간 DEG
deg_cluster <- FindMarkers(
obj,
ident.1 = "T cells",
ident.2 = "B cells",
min.pct = 0.25,
logfc.threshold = 0.25
)
# 2. 상위 DEG 확인
deg_cluster %>%
rownames_to_column("gene") %>%
arrange(desc(avg_log2FC)) %>%
head(20)
# 3. heatmap
top_genes <- rownames(deg_cluster)[1:20]
DoHeatmap(obj, features = top_genes) + NoLegend()
# 4. violin plot
VlnPlot(obj, features = c("CD3D", "MS4A1", "EPCAM"), group.by = "celltype", ncol = 3)
# 5. enrichment
sig_genes <- deg_cluster %>%
rownames_to_column("gene") %>%
filter(p_val_adj < 0.05, avg_log2FC > 0.5) %>%
pull(gene)
gene_df <- bitr(sig_genes,
fromType = "SYMBOL",
toType = "ENTREZID",
OrgDb = org.Hs.eg.db)
ego <- enrichGO(
gene = gene_df$ENTREZID,
OrgDb = org.Hs.eg.db,
ont = "BP",
pAdjustMethod = "BH",
readable = TRUE
)
dotplot(ego)
- DEG 분석은 cluster 간 또는 condition 간 차이를 유전자 수준에서 확인하는 과정입니다.
FindMarkers()를 이용해 두 그룹을 비교할 수 있습니다.- Heatmap, volcano plot, violin plot으로 DEG 결과를 시각화할 수 있습니다.
- GO/KEGG/GSEA 분석을 통해 biological pathway 수준의 해석이 가능합니다.